Transforming Acute Pain Management in Sickle Cell Disease: Where Are We Now?
Introduction
Sickle cell disease (SCD) affects millions worldwide and has become a major public health dilemma in the United States. Although the national incidence is unknown, an estimated 100,000 people are believed to be affected.[1] In 2004, there were approximately 113,000 hospitalizations for sickle cell–related illnesses in the United States, representing an estimated $488 billion in healthcare expenditures.[2]
The future of pain management for this patient group is promising... [this could] improve morbidity, decrease hospital stays, and decrease hospitalizations.
SCD comprises a group of inherited hemoglobinopathies that cause characteristic sickling of red blood cells. Hallmarks of the disease include vaso-occlusion, chronic hemolysis, and increased adhesion of cells to the vascular endothelium, which can lead
to multiorgan dysfunction and early mortality. Specifically, vaso-occlusion results in microvascular obstruction, ischemia, and tissue damage at various anatomical sites, which most commonly manifest as severe recurrent episodes of acute pain (Figure
1). Increased inflammation and alterations in nociception also play a role. The frequency, occurrence, and severity of acute pain episodes vary greatly among individuals with the disease, and patients may choose to manage episodes at home using an
individualized care plan or seek medical attention. Here, we focus on inpatient management of acute pain episodes in patients with SCD.
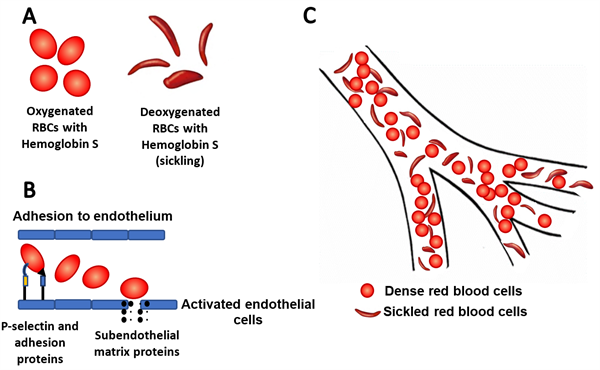
Figure 1A: Sickling of cells with abnormal hemoglobin S during deoxygenation. 1B: The up-regulation of P-selectin and other adhesion molecules in endothelial cells and platelets leads to abnormal
rolling, slow flow and adhesion of sickle cells to vessel surfaces. 1C: Vaso-occlusion of microvasculature by red blood cells.
Overview of Acute Pain Management
Patients who present to the emergency department or a day hospital should have rapid assessment of pain and other SCD-related comorbidities that may require treatment. Early assessment and aggressive management are paramount. Key components of the clinical assessment include the patient’s report of the following.
- Pain onset
- Location
- Quality of pain
- Intensity of pain
- Similarity with prior episodes
- Associated symptoms
- Outpatient analgesic use
- Known effective agents and doses
- Past experience with side effects
Analgesia should be provided promptly, and treatment efficacy should be assessed frequently. Individuals whose pain is not adequately treated at a day hospital or in the emergency room should be admitted to the hospital to escalate therapy.[3] Primary management is pain control; however, hydration and venous thromboembolism prophylaxis should not be overlooked. Below, we summarize the available therapeutic options for acute pain episodes in SCD.
Opioids
Opioids are the mainstay of treatment for acute pain episodes in patients with SCD.[3-6] They include morphine, hydromorphone, and fentanyl. In most cases, patients present after inadequate pain control at home with short and/or long-acting oral opioids. Thus, intravenous therapy with scheduled dosing or continuous dosing via patient-controlled analgesia is recommended for SCD patients admitted for pain control.[7],[8] Several challenges exist with the frequent use of opioid therapy, particularly that of opioid tolerance and opioid-induced hyperalgesia due to N-methyl-D-aspartate (NMDA) receptor activation. Tolerance results in escalating dosage requirements over time, while hyperalgesia may require tapering opioids and a change in therapy.
Other Treatments
Several studies have shown promising results for non-opioid therapies as adjuncts to treatment in patients refractory to opioids. The mechanism of action of these agents are summarized in Figure 2.
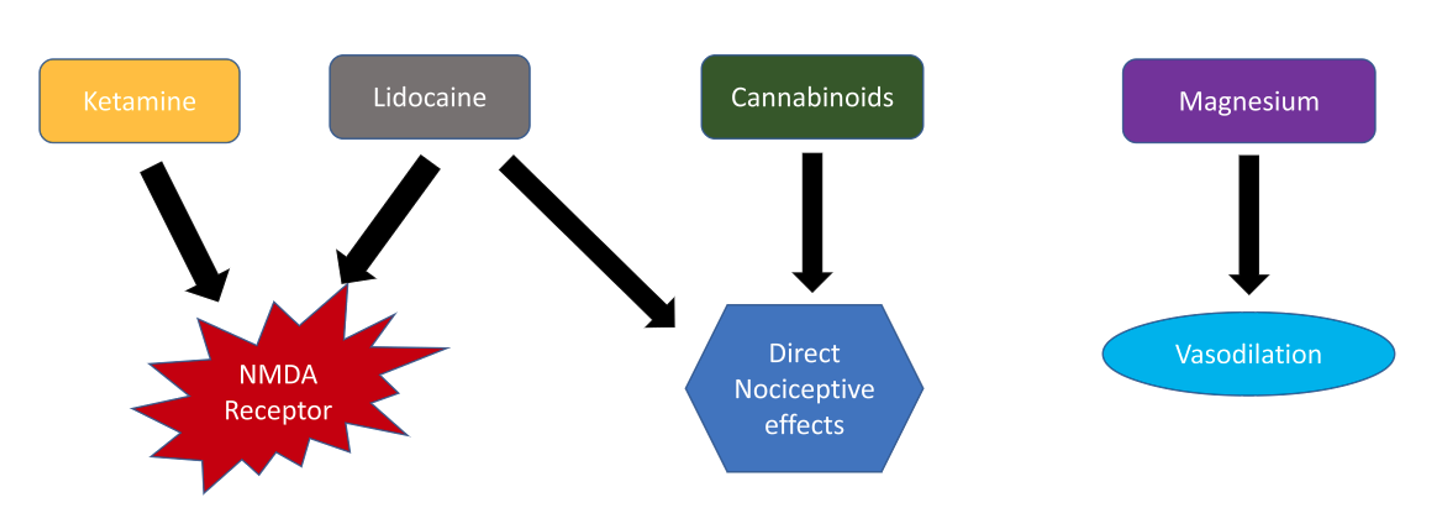 Figure 2: Summary of possible treatment modalities for the management of pain in vaso-occlusive crisis.
Figure 2: Summary of possible treatment modalities for the management of pain in vaso-occlusive crisis.
Ketamine
Ketamine is a noncompetitive antagonist at the NMDA receptor, which has been shown to modulate opioid tolerance and opioid-induced hyperalgesia. It also has anti-inflammatory properties[8] that may be specifically useful during acute pain episodes in SCD. Low (subanesthetic) doses of ketamine are considered a safe and useful adjuvant to opioid analgesia.[9],[10] One study found that a rate of 0.1–0.3 mg/kg/h is sufficient; however, patients with high opioid tolerance and intractable pain may require doses as high as 0.5 mg/kg/h.[10] Cardiovascular, respiratory, and neurologic monitoring by the bedside nurse is required during the infusion and for 1 hour after its completion.
Lidocaine
Lidocaine is an amide local anesthetic that inhibits NMDA and G protein-coupled receptors. In addition to analgesic properties, systemic lidocaine provides both anti-hyperalgesic and anti-inflammatory properties, which may help treat acute pain episodes refractory to opioids. Systemic lidocaine also provides relief of neuropathic pain, a major factor contributing to acute pain in SCD patients.[11]
Magnesium
In some studies, magnesium also has been shown to be effective in acute pain episodes when used in conjunction with standard treatments at a dose of 40 mg/kg (maximum dose 2.5 g) every 8 hours in the pediatric population.[12] Some researchers postulate that the use of magnesium may be beneficial in SCD due to its vasodilatory action.[13] An effective dose in the adult population has not been ascertained, and no studies have shown a reduction of hospital length of stay in adults.[14]
Cannabinoids
Cannabinoids are active in the central nervous system and have direct effects on nociceptive pain. Therefore, cannabinoid receptor agonists may provide beneficial therapeutic effects in addition to opioids during acute pain episodes of SCD. A synergistic interaction between opioids and cannabinoid systems has been described; however, further research is required to ascertain these potentials.[15] These agents are thought to reduce systemic inflammation while targeting cannabinoid receptors in the central nervous system and additional opioid receptors called nociceptin receptors.[16] An ongoing clinical trial seeks to determine the effect of vaporized cannabis on pain and circulating inflammatory and nociceptive markers in patients with SCD (NCT01771731).
Non-Pharmacologic Methods
Non-pharmacologic methods can be used as adjuncts to therapy. These include the following.
- Yoga[17]
- Transcutaneous electrical nerve stimulation
- Warm compression
- Acupuncture
- Emotional support
- Cognitive methods such as deep breathing exercises, music therapy, distraction and cognitive behavioral therapy[12]
Prevention of Acute Pain Episodes
Crizanlizumab
Crizanlizumab is a humanized monoclonal antibody against the adhesion molecule P-selectin. The up-regulation of P-selectin in endothelial cells and platelets contributes to erythrocyte and leukocyte adhesion to vessel walls and subsequent vaso-occlusion (Figure 1B). In a recent randomized clinical trial, treatment with high-dose crizanlizumab resulted in an 45.3% lower annual rate of sickle cell-related pain crises over placebo. The median times to the first and second acute pain episode were two to three times as long in patients taking high dose crizanlizumab over placebo.[18]
L-Glutamine
Sickled red blood cells have increased levels of reactive oxygen species, and amino acid L-glutamine is needed to reduce oxidative stress. This forms the basis of a phase III randomized trial of L-glutamine powder that showed a reduced number of pain crises over 48 weeks in patients who received l-glutamine, regardless of hydroxyurea use. [19]
Barriers to Care
Several studies have found that negative provider attitudes and a lack of knowledge of the standard of care contribute to barriers to effective management of acute pain episodes.[20] Certain aspects of managing opioid complications are unique to individuals with SCD due to the lifelong unremitting nature of the pain and chronic requirement for opioids in some individuals, resulting in opioid tolerance. Patients then require higher doses for the same effect. Many healthcare providers fear the adverse effects of opioids, particularly sedation and respiratory failure, and want to avoid opioid abuse and diversion. This often presents barriers to adequate pain management including disbelief of pain reports,[21],[22] reluctance to prescribe therapy, and insufficient treatment.[23] Unfortunately, there are no definitive ways to identify drug-seeking patients. However, multiple tools can assist in decision-making by identifying aberrant behavior related to opioid misuse, including the following.
- Web-based prescription monitoring programs allow providers to view all the patient’s opioid prescriptions and evaluate aberrancies such as frequent emergency department visits, use of multiple providers, and simultaneously active opioid prescriptions.
- Urine toxicology both historically and at the time of diagnosis can determine whether the patient is taking their medications or other illicit substances.
- Self-administered screening tools can be used in the non-acute setting to identify and monitor aberrant behavior (eg, the Screener and Opioid Assessment for Patients with Pain [SOAPP or SOAPP-R] and the Current Opioid Misuse Measure [COMM or COMM-9]).[24-26]
Nonetheless, there is no evidence to support the misconception that the use of opioids is associated with in-hospital mortality among SCD patients in the United States.[27] Individuals may also present atypically and may not even appear distressed, leading to suspicion of drug-seeking behavior. Because of the episodic nature of pain in these patients, they may present for medical attention multiple times per year and be labeled as “frequent flyers.” Distrust between health care providers and patients with acute pain episodes may result. This is compounded by the perception that SCD patients have higher rates of opioid addiction.[28-30] However, to date, there is no evidence to support this belief.[30]
Conclusion
SCD pain is multifactorial and can be acute, chronic, or acute on chronic with numerous barriers to effective management, making treatment of acute sickle crisis extremely challenging. Opioids remain the mainstay of pain management, but this is not without adverse effects. While some adjuncts to therapy have been studied, research is still ongoing. The FDA also has approved preventive agents. All in all, the future of pain management for this patient group is promising. This could lead not only to the transformation of the standard of care but also improve morbidity, decrease hospital stays, and decrease hospitalizations.
References
- Centers for Disease Control and Prevention. Data & statistics on sickle cell disease. https://www.cdc.gov/ncbddd/sicklecell/data.html. Published August 31, 2016. Accessed January 5, 2020.
- Steiner CA, Miller JL. Sickle cell disease patients in U.S. hospitals, 2004: statistical brief #21. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD: Agency for Healthcare Research and Quality (US); 2006. Available at: http://www.ncbi.nlm.nih.gov/books/NBK63489/. Accessed November 22, 2019.
- Brookoff D, Polomano R. Treating sickle cell pain like cancer pain. Ann Intern Med. 1992;116(5):364-8. https://doi.org/10.7326/0003-4819-116-5-364
- Uzun B, Kekec Z, Gurkan E. Efficacy of tramadol vs meperidine in vasoocclusive sickle cell crisis. Am J Emerg Med. 2010;28(4):445-9. https://doi.org/10.1016/j.ajem.2009.01.016
- Gonzalez ER, Ornato JP, Ware D, Bull D, Evens RP. Comparison of intramuscular analgesic activity of butorphanol and morphine in patients with sickle cell disease. Ann Emerg Med. 1988;17(8):788-91. https://doi.org/10.1016/s0196-0644(88)80554-7
- Jacobson SJ, Kopecky EA, Joshi P, Babul N. Randomised trial of oral morphine for painful episodes of sickle-cell disease in children. Lancet. 1997;350(9088):1358-61. https://doi.org/10.1016/S0140-6736(97)08462-6
- Udezue E, Herrera E. Pain management in adult acute sickle cell pain crisis: a viewpoint. West Afr J Med. 2007;26(3):179-82. https://doi.org/10.4314/wajm.v26i3.28305
- Cho JE, Shim JK, Choi YS, Kim DH, Hong SW, Kwak YL. Effect of low-dose ketamine on inflammatory response in off-pump coronary artery bypass graft surgery. Br J Anaesth. 2009;102(1):23-8. https://doi.org/10.1093/bja/aen325
- Uprety D, Baber A, Foy M. Ketamine infusion for sickle cell pain crisis refractory to opioids: a case report and review of literature. Ann Hematol. 2014;93(5):769–71. https://doi.org/10.1007/s00277-013-1954-3
- Puri L, Morgan KJ, Anghelescu DL. Ketamine and lidocaine infusions decrease opioid consumption during vaso-occlusive crisis in adolescents with sickle cell disease. Curr Opin Support Palliat Care. 2019;13(4):402–7. https://doi.org/10.1097/SPC.0000000000000437
- Nguyen NL, Kome AM, Lowe DK, Coyne P, Hawks KG. Intravenous lidocaine as an adjuvant for pain associated with sickle cell disease. J Pain Palliat Care Pharmacother. 2015;29(4):359–64. https://doi.org/10.3109/15360288.2015.1082009
- Brousseau DC, Scott JP, Hillery CA, Panepinto JA. The effect of magnesium on length of stay for pediatric sickle cell crisis. Acad Emerg Med. 2004;11(9):968–72. https://doi.org/10.1197/j.aem.2004.04.009
- Uwaezuoke SN, Ayuk AC, Ndu IK, Eneh CI, Mbanefo NR, Ezenwosu OU. Vaso-occlusive crisis in sickle cell disease: current paradigm on pain management. J Pain Res. 2018;11:3141–50. https://doi.org/10.2147/JPR.S185582
- Than NN, Soe HHK, Palaniappan SK, Abas AB, De Franceschi L. Magnesium for treating sickle cell disease. Cochrane Database Syst Rev. 2017;4(4):CD011358. https://doi.org/10.1002/14651858.CD011358.pub2
- Howard J, Anie KA, Holdcroft A, Korn S, Davies SC. Cannabis use in sickle cell disease: a questionnaire study. Br J Haematol. 2005;131(1):123–8. https://doi.org/10.1111/j.1365-2141.2005.05723.x
- Tran H, Gupta M, Gupta K. Targeting novel mechanisms of pain in sickle cell disease. Blood. 2017;130(22):2237-85. https://doi.org/10.1182/blood-2017-05-782003
- Moody K, Abrahams B, Baker R, et al. A randomized trial of yoga for children hospitalized with sickle cell vaso-occlusive crisis. J Pain and Symptom Manage. 2017;53(6):1026-34. https://doi.org/10.1016/j.jpainsymman.2016.12.351
- Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-39. https://doi.org/10.1056/NEJMoa1611770
- Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226-35. https://doi.org/10.1056/NEJMoa1715971
- Brennan-Cook J, Bonnabeau E, Aponte R, Augustin C, Tanabe P. Barriers to care for persons with sickle cell disease: the case manager's opportunity to improve patient outcomes. Prof Case Manag. 2018;23(4):213–19. https://doi.org/10.1097/NCM.0000000000000260
- Goddu AP, O’Conor KJ, Lanzkron S, et al. Do words matter? Stigmatizing language and the transmission of bias in the medical record. J Gen Intern Med. 2018;33(5):685-91. https://doi.org/10.1007/s11606-017-4289-2
- Labbé E, Herbert D, Haynes J. Physicians’ attitude and practices in sickle cell disease pain management. J Palliat Care. 2005;21(4):246-51. https://doi.org/10.1177/082585970502100403
- Ballas SK. The sickle cell painful crisis in adults: phases and objective signs. Hemoglobin. 1995;19(6):323-33. https://doi.org/10.3109/03630269509005824
- Black RA, McCaffrey SA, Villapiano AJ, Jamison RN, Butler SF. Development and validation of an eight-item brief form of the SOAPP-R (SOAPP-8). Pain Med. 2018;19(10):1982-7. https://doi.org/10.1093/pm/pnx194
- Butler SF, Budman SH, Fernandez KC, et al. Development and validation of the current opioid misuse measure. Pain. 2007;130(1-2):144-56. https://doi.org/10.1016/j.pain.2007.01.014
- McCaffrey SA, Black RA, Villapiano AJ, Jamison RN, Butler SF. Development of a brief version of the current opioid misuse measure (COMM): the COMM-9. Pain Med. 2019;20(1):113-8. https://doi.org/10.1093/pm/pnx311
- Akinboro OA, Nwabudike S, Edwards C, et al. Opioid use is NOT associated with in-hospital mortality among patients with sickle CELL disease in the United States: findings from the national inpatient sample. Blood. 2018;132(Supplement 1):315. https://doi.org/10.1182/blood-2018-99-115573
- Shapiro BS, Benjamin LJ, Payne R, Heidrich G. Sickle cell-related pain: perceptions of medical practitioners. J Pain Symptom Manage. 1997;14(3):168-74. https://doi.org/10.1016/S0885-3924(97)00019-5
- Pack-Mabien A, Labbe E, Herbert D, Haynes J. Nurses’ attitudes and practices in sickle cell pain management. Appl Nurs Res. 2001;14(4):187-92. https://doi.org/10.1053/apnr.2001.26783
- Ruta NS, Ballas SK. The opioid drug epidemic and sickle cell disease: guilt by association. Pain Med. 2016;17(10):1793-8. https://doi.org/10.1093/pm/pnw074